Objective | To use bayesian methods to explore the interdependence and correlations between bond charge correction parameters and to quantify the performance of difference combinations of atom types. |
Key outcomes | List expected outcomes and success metrics |
Status | 
|
NOTE - This is just an initial brain dump so far. Owen Madin will refine this into a full study.
Problem Statement
AM1BCC parameters have been converted to a SMIRNOFF compatible format:

In general a BCC parameter is constructed by combinatorially concatenating two atom environments (defined by separate SMIRKS patterns) with a bond environment (single, double, delocalised, also defined by a SMIRKS pattern).
Ideally we would like to refit these against at least QM electrostatic potential (ESP) data.
There are many different, and in some cases very specific atom and bond types which don’t necessarily map well into the SMIRKS language, e.g. (highly) delocalised lone pairs on nitrogens and dative bonds. Further, there are a total of 354 types - are all of these truly justified by the data we are trying to reproduce?
Bayesian methods give access to posterior distributions from which the correlations (including more complex, non-linear relationships) between BCC parameters can be easily discerned. Further the computation of Bayes factors allow us to quantitatively measure where extra model complexity (in this case extra ‘atom’ types or BCC ‘types’). Combined, Bayes methods should allow us to gain data driven insight into where we perhaps have too many atom types, and where there are types missing.
In particular, N sets of different atom types could be proposed (e.g. one set may have the highly delocalised and delocalised N merged into a single atom type), their Bayes factor computed, and inference made off of that as to which the ESP data supports the most.
(Possible extension to this project - can we just define atom types in terms of an element and it’s connectivity, and then use Wiberg bond order interpolation to scale the BCC? Is there a way to directly incorporate the Wiberg bond order when deciding which type a bond is in? I.e. easily define ‘delocalised' bonds?)
Scope
Start with C, O, H, (reasonably low dimensionality), then extend to N and S (much higher dimensionality).
Train on a least some molecules ideally taken from something like the NCI list or enamine REAL
Methodology
The optimisation of the BCC parameters against ESP data can be written as a Bayesian linear regression problem (or at least, using the same form for the loss function).
A given set of bond charge correction parameters
\vec{p} =
\begin{bmatrix}
p_1 \\
\vdots \\
p_{\gamma} \\
\end{bmatrix} |
can be applied to a particular molecule using an assignment matrix
T=\begin{bmatrix}
T_{1,1} & \dots & T_{1,\gamma} \\
\vdots & \ddots & \vdots \\
T_{N,1} & \dots. & T_{N,\gamma} \\
\end{bmatrix} |
such that
\vec{q}_{corr} =
\begin{bmatrix}
T_{1,1} & \dots & T_{1,\gamma} \\
\vdots & \ddots & \vdots \\
T_{N,1} & \dots. & T_{N,\gamma} \\
\end{bmatrix}
\begin{bmatrix}
p_1 \\
\vdots \\
p_{\gamma} \\
\end{bmatrix} |
where 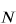 is the number of atoms in the molecule and
is the number of atoms in the molecule and 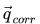 is the x 1 vector of partial charge corrections to apply to each atom.
is the x 1 vector of partial charge corrections to apply to each atom.
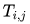 represents the number of times that BCC parameter
represents the number of times that BCC parameter 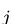 should be applied to atom
should be applied to atom 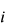 . E.g. in the case of methanol which would only need a single BCC parameter
. E.g. in the case of methanol which would only need a single BCC parameter
\vec{q}_{corr} =
\begin{bmatrix}
4 \\
-1 \\
-1 \\
-1 \\
-1 \\
\end{bmatrix}
\begin{bmatrix}
\vec{p}_{CH} \\
\end{bmatrix} |
The total partial charges 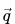 on the molecule are then
on the molecule are then
\vec{q} = \vec{q}_{qm} + \vec{q}_{corr} |
where 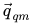 are a set of partial charges computed directly from a QM method such as AM1. The ESP
are a set of partial charges computed directly from a QM method such as AM1. The ESP 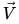 at a set of
at a set of 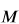 grid points can be computed as
grid points can be computed as
\vec{V} =
\begin{bmatrix}
\dfrac{1}{r_{1,1}} & \dots & \dfrac{1}{r_{1,N}} \\
\vdots & \ddots & \vdots \\
\dfrac{1}{r_{M,1}} & \dots. & \dfrac{1}{r_{M,N}} \\
\end{bmatrix}
\begin{bmatrix}
\vec{q}_1 \\
\vdots \\
\vec{q}_N \\
\end{bmatrix} |
where 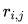 is the distance between grid point and atom .
is the distance between grid point and atom .
can be split into the contributions from the QM charges and the charge corrections:
where 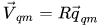 and
and
V_{corr} =
\begin{bmatrix}
\dfrac{1}{r_{1,1}} & \dots & \dfrac{1}{r_{1,N}} \\
\vdots & \ddots & \vdots \\
\dfrac{1}{r_{M,1}} & \dots. & \dfrac{1}{r_{M,N}} \\
\end{bmatrix}
\begin{bmatrix}
T_{1,1} & \dots & T_{1,\gamma} \\
\vdots & \ddots & \vdots \\
T_{N,1} & \dots. & T_{N,\gamma} \\
\end{bmatrix}
\begin{bmatrix}
p_1 \\
\vdots \\
p_{\gamma} \\
\end{bmatrix} |
Here we denote
X_i=
\begin{bmatrix}
\dfrac{1}{r_{1,1}} & \dots & \dfrac{1}{r_{1,N}} \\
\vdots & \ddots & \vdots \\
\dfrac{1}{r_{M,1}} & \dots. & \dfrac{1}{r_{M,N}} \\
\end{bmatrix}
\begin{bmatrix}
T_{1,1} & \dots & T_{1,\gamma} \\
\vdots & \ddots & \vdots \\
T_{N,1} & \dots. & T_{N,\gamma} \\
\end{bmatrix} |
as the design matrix for molecule (or conformer)
Assuming a normal likelihood, the Bayesian likelihood function then becomes
\rho \left(\vec{V}_{diff} | X_i, p, \sigma^2\right) \propto \left(\sigma^2\right)^{-n/2} \exp \left(-\dfrac{1}{2\sigma^2} \left(\vec{V}_{diff} - X_i\vec{p}\right)^T\left(\vec{V}_{diff} - X_i\vec{p}\right)\right) |
where
\vec{V}_{diff} = \vec{V}_{target,i} - \vec{V}_{qm} |
and 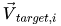 is an M x 1 vector of the target ESP data for molecule .
is an M x 1 vector of the target ESP data for molecule .
For K molecules / conformers
X = \begin{bmatrix}
X_1 \\
\vdots \\
X_K \\
\end{bmatrix} |
and
\vec{V}_{target} = \begin{bmatrix}
\vec{V}_{target, 1} \\
\vdots \\
\vec{V}_{target, K} \\
\end{bmatrix} |
None of 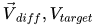 or
or 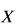 depend upon the values of
depend upon the values of 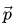 and so can be pre-computed before the optimisation making the likelihood function rapid to evaluate.
and so can be pre-computed before the optimisation making the likelihood function rapid to evaluate.
Example implementations are given here:

Choice of Priors
Priors need to be chosen for each of the BCC parameters, as well as for sigma.
For sigma most literature seems to suggest something weakly informative like a half cauchy or a half student T.
For the parameters a Normal distribution with mean 0 and STD 1 would seem to make sense. Given that the base QM method should capture the salient features of the electronic charge distribution, especially the formal charge distribution the values are expected to be either positive or negative, and and likely less than 0.5 in magnitude.
Bayes Factors
These could either be computed by performing RJMC, but could also be computed using ‘free-energy’ like methods such as MBAR whereby a lambda value is introduced which transforms an analytically tractable distribution into the full posterior distribution.
The ‘free-energy’ approach may be better here - this should be easily implementable in current frameworks such as Pyro, and would also mean not having to worry about designing the transition matrices.
ESP
The ESP data will be computed on a FCC grid (spacing TBD) and using a aug-cc-pV(D+d)Z basis and the pw6b95 method as was highlighted by the RESP2 publication as yielding a strong balance of performance and accuracy.